
IB Syllabus focus:
'- Understand the molecular clock method.
- Learn to use base sequences to construct cladograms.'
The quest to decipher the chronology and relationships of species' evolution has seen the birth of some fascinating tools. Among them, the molecular clock and cladograms have illuminated evolutionary paths with a precision rooted in genetics.
The Molecular Clock Method
Evolution, as it turns out, has a predictable rhythm when observed at the molecular level. The molecular clock method endeavours to measure this rhythm.
The Fundamentals of Molecular Clocks
Molecular Clock: A technique that estimates the time of evolutionary divergence between species by comparing the number of genetic mutations, assuming a relatively constant rate of molecular change over time.
Concept of Constancy: At its core, the molecular clock hypothesis hinges on the idea that genes or protein regions evolve at a consistent rate. If this evolutionary rate is known, differences between two species can be assessed to approximate when they branched apart.
Neutral Theory: A pivotal concept introduced by Motoo Kimura, the neutral theory posits that a significant portion of evolutionary molecular changes neither advantages nor disadvantages an organism. This forms the bedrock for the molecular clock's predictive powers.Molecular clock hypothesis
Image courtesy of Understanding Evolution
Calibration: Anchoring the Clock
Linking to the Fossil Record: For the molecular clock to be precise, its readings must be calibrated. Fossil records or known evolutionary events often offer this calibration. For example, if fossils reveal two species parted ways 10 million years ago and their genetic code has 20 distinct mutations, then one could deduce a rate of 2 mutations every million years.
Recognising the Clock's Limitations
Variability in Mutation Rates: Not all genes or proteins evolve at identical rates. External factors or environmental pressures can accelerate or decelerate these rates, making some readings variable.
The Pitfall of Overlapping Mutations: In lineages stretching back eons, it's possible for mutations to occur at the same genetic site multiple times. This can cloud readings and underestimate the actual time of divergence.
Cladograms and Base Sequences: Plotting Relationships
Cladogram: A branching diagram that illustrates the evolutionary relationships among different species or groups, showing points at which they diverged from common ancestors.
Cladograms, with their branching narratives, beautifully depict relationships among organisms. In modern times, molecular biology has enriched these diagrams with DNA and RNA sequence data.
Constructing Cladograms: A Step-by-step Approach
Gathering Sequences: The process begins with obtaining DNA or RNA sequences from the organisms in focus.
Aligning the Sequences: Advanced bioinformatics tools facilitate the alignment of these sequences. This step highlights regions of similarity and difference, offering initial insights into relatedness.
Computing Differences: The number of disparities (or mutations) between organisms are tabulated. A smaller number signifies a closer relationship and vice versa.
Genetic Distance (D) = Number of Differences (n) ÷ Total Number of Base Pairs Compared (N)
D (unitless) = Measure of genetic divergence
n (count) = Number of nucleotide differences observed between two sequences
N (count) = Total number of nucleotide sites compared
Drafting the Cladogram:
Constructing a Distance Matrix: This matrix is a visual representation where each cell indicates the genetic disparity between two organisms.
Charting Branching Patterns: Using the matrix, probable branching sequences are determined. Organisms with fewer differences will be closer neighbours on the cladogram.
Cladogram Interpretation: Unearthing Stories
How to Navigate Cladograms: The foundational ancestor is represented by the cladogram's root. Each divergence point (node) signals a speciation event in evolutionary history.
The Message in Branch Lengths: In some instances, branch lengths reflect the degree of genetic change, providing additional layers of understanding.
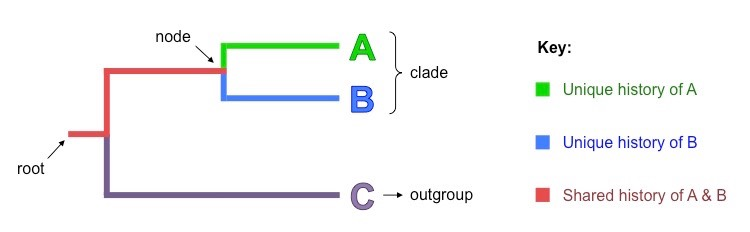
Image courtesy of BioNinja
The Value of Molecular Cladograms
Unbiased Data Source: While physical traits can sometimes mislead due to phenomena like convergent evolution, DNA and RNA sequences offer objective, unbiased data.
Universality in Measurement: Since all life forms possess DNA or RNA, it's a universally applicable metric, facilitating comparisons across a vast spectrum of organisms.
Revealing Hidden Links: Beyond physical appearances, molecular cladistics can unveil deep-seated relationships, granting a richer perspective on evolution.
Challenges in Harnessing Molecular Data
The Challenge of Homoplasy: Similar to physical traits, molecular data can exhibit homoplasy. Traits might emerge in separate lineages, either due to convergent evolution or genetic reversions.
Homoplasy: The independent appearance of similar traits or genetic sequences in different evolutionary lineages, not due to common ancestry but often caused by convergent evolution or genetic reversal.
Dealing with Gene Duplication: A gene can sometimes replicate itself, producing multiple copies within the same organism. This duplication can introduce ambiguities when discerning evolutionary relationships.
Navigating Horizontal Gene Transfer: Particularly among microbes, genes can traverse between unrelated organisms. Such transfers can muddle the clear waters of lineage tracing.
FAQ
Horizontal gene transfer (HGT) is the direct transfer of genetic material between unrelated organisms, typically observed among bacteria. This non-traditional form of gene transfer can confound molecular cladistics because it introduces genetic material from an external source, making it difficult to trace a linear path of inheritance. When HGT occurs, genes in an organism may have different evolutionary histories, meaning they don't all trace back to a singular common ancestor. This can complicate or even mislead efforts to construct an accurate cladogram based solely on molecular data, emphasizing the need for comprehensive approaches in evolutionary biology studies.
The root of a cladogram represents the most ancestral branch or the common ancestor of all the taxa displayed in the diagram. It's the starting point from which all species or groups diverged. Determining the root is crucial for understanding the directional flow of evolutionary changes. The root is usually identified using an outgroup, which is a species or group known to be closely related but not part of the studied groups. By comparing the outgroup's characteristics with those of the studied groups, researchers can infer which traits are ancestral and which are derived, allowing them to appropriately position the root.
Bioinformatics tools play a pivotal role in handling and analysing the vast amounts of genetic data needed for cladogram construction. Firstly, they assist in aligning genetic sequences from different organisms, highlighting similarities and differences. This alignment helps researchers pinpoint mutations and quantify the genetic distance between species. Furthermore, sophisticated algorithms can automatically generate distance matrices and propose likely branching patterns based on computed genetic differences. Without these tools, the manual analysis of extensive genetic data would be impractical, making bioinformatics essential for modern molecular cladistics.
Genetic reversions refer to the phenomenon where a mutation undergoes a reversal, returning to its original state. In the context of the molecular clock, this means a previously counted mutation could be erased, potentially leading to an underestimation of the time since divergence. If not accounted for, this can mislead researchers into believing that two species shared a more recent common ancestor than they truly did. Genetic reversions, while relatively rare, underscore the importance of using multiple genes or regions to calibrate and validate the molecular clock to ensure accurate evolutionary timelines.
Certain genes evolve at different rates due to their varied roles and the selective pressures acting upon them. However, the molecular clock concept doesn't suggest that all genes have the same rate of mutation, but rather that specific genes or protein regions can evolve at a consistent rate. When using the molecular clock method, it's essential to select genes or regions that are known for their consistent evolutionary rates, typically those under minimal selective pressure. Additionally, by comparing multiple genes or proteins and averaging their mutation rates, scientists can obtain a more reliable estimate of divergence times.
Practice Questions
The molecular clock method is based on the principle that certain genes or protein regions evolve at a relatively constant rate over time. If this rate of mutation is known, by observing the differences or mutations in the genetic code of two species, scientists can calculate an approximate time of when these species diverged. Calibration is vital to enhance the accuracy of this method. To calibrate the molecular clock, fossil records are employed. If fossils suggest two species diverged at a specific time in the past, and their genes exhibit a known number of mutations, then this information can be used to establish a rate of mutation, thereby calibrating the molecular clock for more precise future readings.
Cladograms are branching diagrams showcasing relationships between organisms based on shared characteristics. When constructing cladograms using base sequences, the first step involves collecting DNA or RNA sequences of the organisms in question. Next, these sequences are aligned using bioinformatics tools to identify similarities and differences. The third step is counting the differences between each pair of organisms, allowing for an assessment of their relatedness. The data is then used to create a distance matrix, representing genetic disparities between organisms. Using this matrix, branching patterns are determined for the cladogram's construction. One challenge in using molecular data is the occurrence of homoplasy, where a trait might appear in separate lineages, often due to convergent evolution or genetic reversions, making it challenging to determine true evolutionary relationships.
